Search results
Search for "machine learning" in Full Text gives 19 result(s) in Beilstein Journal of Organic Chemistry.
GlAIcomics: a deep neural network classifier for spectroscopy-augmented mass spectrometric glycans data
Beilstein J. Org. Chem. 2023, 19, 1825–1831, doi:10.3762/bjoc.19.134

- analysis, thus excluding manual interpretation. Besides, in the prospective of deploying the technology beyond the molecular spectroscopy community, it is essential to develop an automated, reliable, and robust strategy for the analysis of the spectroscopic data. Machine learning methods appear to be
- spectra for cancer classification [8] and many research groups focused their efforts on using machine learning for simulating molecular structures; generating vibrational spectra; and classifying chemical groups based on vibrational features [9][10]. In a recent publication, the random forest approach was
- proposed to identify the presence of structural features in oligosaccharides based on their gas-phase IR spectra [11]. To the best of our knowledge, machine learning classification studies have not been reported to identify saccharides using MS–IR carbohydrate analysis. Here, we report a study of a






Functional characterisation of twelve terpene synthases from actinobacteria
Beilstein J. Org. Chem. 2023, 19, 1386–1398, doi:10.3762/bjoc.19.100

- for a pathway reconstruction towards artemisinin. The increased knowledge about terpene synthases together with the structures of their products will also be of interest for machine learning approaches to enable the prediction of terpene synthase functions from their amino acid sequences. Both aspects










Bromination of endo-7-norbornene derivatives revisited: failure of a computational NMR method in elucidating the configuration of an organic structure
Beilstein J. Org. Chem. 2023, 19, 764–770, doi:10.3762/bjoc.19.56

- Kutateladze claimed that based on an applied machine learning-augmented DFT method for computational NMR that the structure of the product, (1R,2R,3S,4S,7s)-2,3,7-tribromobicyclo[2.2.1]heptane was wrong. With the aid of their computational method, they revised a number of published structures, including ours
- have developed a machine learning-augmented DFT method for computational NMR, DU8ML, for fast and ‘accurate’ computational approaches [2]. They applied this computational method to a number of previously published organic compounds and claimed to have revised some structures and proposed new mechanisms












Total synthesis: an enabling science
Beilstein J. Org. Chem. 2023, 19, 474–476, doi:10.3762/bjoc.19.36

- [12], as illustrated in this thematic issue with the synthesis of pheromones [16]. This requires permanent technological progress. Thus, the recent boom of artificial intelligence, machine learning, and computational chemistry for retrosynthetic analyses and beyond foreshadows a renewed interest in


Navigating and expanding the roadmap of natural product genome mining tools
Beilstein J. Org. Chem. 2022, 18, 1656–1671, doi:10.3762/bjoc.18.178

- developed for the biosynthetic rule-based identification of natural product gene clusters. Apart from these hard-coded algorithms, multiple tools that use machine learning-based approaches have been designed to complement the existing genome mining tool set and focus on natural product gene clusters that
- lack genes with conserved signature sequences. In this perspective, we take a closer look at state-of-the-art genome mining tools that are based on either hard-coded rules or machine learning algorithms, with an emphasis on the confidence of their predictions and potential to identify non-canonical
- algorithms based on hard-coded rules to machine learning (ML)-based approaches with regard to the natural product biosynthetic principles they are most suited for. We focus on how the different genome mining tools identify BGCs and highlight their advantages and limitations. Moreover, we will showcase two








Molecular and macromolecular electrochemistry: synthesis, mechanism, and redox properties
Beilstein J. Org. Chem. 2022, 18, 1505–1506, doi:10.3762/bjoc.18.158

- so on, it has a high affinity to informatics approaches, e.g., machine learning, which is expected to become an increasingly important tool in the future. Progress in the design of organic molecules and polymers and the understanding of the redox behavior of these compounds has led to the development
Cytochrome P450 monooxygenase-mediated tailoring of triterpenoids and steroids in plants
Beilstein J. Org. Chem. 2022, 18, 1289–1310, doi:10.3762/bjoc.18.135

- combination with ground-breaking machine learning approaches for protein structure prediction such as AlphaFold2 [108], we anticipate that the catalytic repertoire of CYPs will be exploited much more for the biotechnological production of tailor-made triterpenoids and steroids in the near future. We hope that







Molecular basis for protein–protein interactions
Beilstein J. Org. Chem. 2021, 17, 1–10, doi:10.3762/bjoc.17.1

- characterisation of the binding reaction. Computational methods are used to predict PPIs and interfaces. The advantage of performing in silico experiments includes narrowing down the number of the binding partners to be tested in vitro or in vivo. Computational methods include supervised machine learning, where





A consensus-based and readable extension of Linear Code for Reaction Rules (LiCoRR)
Beilstein J. Org. Chem. 2020, 16, 2645–2662, doi:10.3762/bjoc.16.215

- such models using a variety of strategies, including mechanistic and nonlinear [4][5][6][7][8][9][10][11][12], linear probabilistic [13][14], machine learning [15], formal-grammar [16], and substructural [17]. Unfortunately, most of these approaches use slightly different expressions of the building



Models of necessity
Beilstein J. Org. Chem. 2020, 16, 1649–1661, doi:10.3762/bjoc.16.137

- not always clear to practicing chemists, so that controversial discussions about the merits of alternative models often arise. However, the extensive use of artificial intelligence (AI) and machine learning (ML) in chemistry, with the aim of being able to make reliable predictions, will require that
- molecules suitable for depiction in databases, cheminformatics, machine learning (ML) or artificial intelligence (AI): It is essential for chemists to be able to communicate with each other about molecules. The language of chemistry varies slightly between the organic and inorganic communities. However, it
- reactions are relatively straightforward constructions, if we look further, for example to systems for predicting reactions or suggesting synthetic routes [45], whether using manually coded transformations or developments using automated machine learning and AI techniques, limitations of the Lewis model





In silico rationalisation of selectivity and reactivity in Pd-catalysed C–H activation reactions
Beilstein J. Org. Chem. 2020, 16, 1465–1475, doi:10.3762/bjoc.16.122
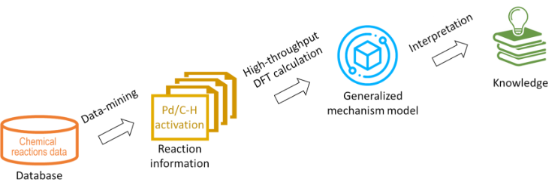
- desired. Recent years have seen the emergence of new methods of research in chemistry and process development, which include high-throughput experiments [3], autonomous self-optimising reactors [4][5][6], as well as predictions of reaction outcomes and of reaction conditions based on machine learning (ML
- demonstrated. While machine learning methods are showing great promise and continue to be improved upon, it is also clear that a ML model is unlikely to ever be able to compete in accuracy and interpretability with fully predictive mechanistic models, were it not for the prohibitively high cost of developing
- developing machine learning models for predicting reaction outcomes. C–H activation reactions allow conversion of relatively inexpensive and abundant hydrocarbons into the more sophisticated value-added molecules [11]. With the notion of step-economical and environmentally friendly synthesis, direct



Photophysics and photochemistry of NIR absorbers derived from cyanines: key to new technologies based on chemistry 4.0
Beilstein J. Org. Chem. 2020, 16, 415–444, doi:10.3762/bjoc.16.40
























Bacterial terpene biosynthesis: challenges and opportunities for pathway engineering
Beilstein J. Org. Chem. 2019, 15, 2889–2906, doi:10.3762/bjoc.15.283

- machine learning and retrobiosynthetic algorithms could facilitate the design of constructs for specific terpenoid variants [149]. While it is now relatively straightforward to direct the flux to produce terpene skeletons, less is known about how to effectively support function of CYPs beyond natural












Steric “attraction”: not by dispersion alone
Beilstein J. Org. Chem. 2018, 14, 1482–1490, doi:10.3762/bjoc.14.125

- functional theory [39], post-Hartree–Fock [40][41], symmetry adapted perturbation theory (SAPT) [42][43][44][45][46] data or to a combination of the latter two (e.g., the monomer electron density force field, MEDFF) [47]. The latter approach has been subsequently exploited in the machine learning






Biomimetic molecular design tools that learn, evolve, and adapt
Beilstein J. Org. Chem. 2017, 13, 1288–1302, doi:10.3762/bjoc.13.125

- , evolving, machine learning-based molecular design and optimization methods are approaching the period of very rapid growth and their impact is already being described as potentially disruptive. This paper describes new developments in biomimetic adaptive, evolving, learning computational molecular design
- methods and their potential impacts in chemistry, engineering, and medicine. Keywords: automated chemical synthesis; deep learning; evolutionary algorithms; in silico evolution; machine learning; materials design and development; neural networks; Introduction There is still not a clear understanding of
- future impact. It introduces the most common type of algorithm, machine learning. A discussion of a very useful machine-learning algorithm, the neural network follows, and problems that often arise in their use, and solutions to these difficulties described. A new type of deep learning neural network















Automating multistep flow synthesis: approach and challenges in integrating chemistry, machines and logic
Beilstein J. Org. Chem. 2017, 13, 960–987, doi:10.3762/bjoc.13.97




















Self-optimisation and model-based design of experiments for developing a C–H activation flow process
Beilstein J. Org. Chem. 2017, 13, 150–163, doi:10.3762/bjoc.13.18

- model-based design of experiments, based on the first principles model structure, in automated flow experiments, and coupling of the process models with a statistical machine learning based target optimisation. We demonstrate that MBDoE offers a significant potential for efficient and rapid generation
- difficulties regarding multi-objective global optimisation can be overcome. Furthermore, the proposed optimisation procedure can deal with potential uncertainties and restricted validity in the physical model. This is achieved by the machine learning functionalities of the MOAL algorithm, which retrain the








Computational methods in drug discovery
Beilstein J. Org. Chem. 2016, 12, 2694–2718, doi:10.3762/bjoc.12.267

- ; machine learning; pharmacophore; QSAR; SBDD; scoring; target flexibility; Introduction Bringing a pharmaceutical drug to the market is a long term process that costs billions of dollars. In 2014, the Tufts Center for the Study of Drug Development estimated that the cost associated with developing and
- generates sequence–template alignments for a query sequence and identifies best structure matches from the PDB [53]. In addition to sequence profile alignments, it also uses multiple structure information as well. DescFold is another webserver which employs SVM-based machine learning algorithms in protein










The Beilstein Journal of Organic Chemistry and the changing face of scientific publishing
Beilstein J. Org. Chem. 2015, 11, 2242–2244, doi:10.3762/bjoc.11.242
- , efficiency of peer review and publishing. Text and data mining, big data and machine learning, will also become routinely possible, but will only become really useful if the scientific community starts storing and making all verified results – including the negative – publically available. In organic